Синдром Прадера-Вилли – врожденное заболевание, при котором возникает сочетание ожирения, низкого роста, снижения функции половых желез (гипогонадизм) и низкого интеллекта. Это заболевание имеет очень широкий спектр проявлений и признаков. Течение болезни отличается в каждом отдельном случае и может варьировать от легкой формы до тяжелой, которая прогрессирует в течение всей жизни человека.
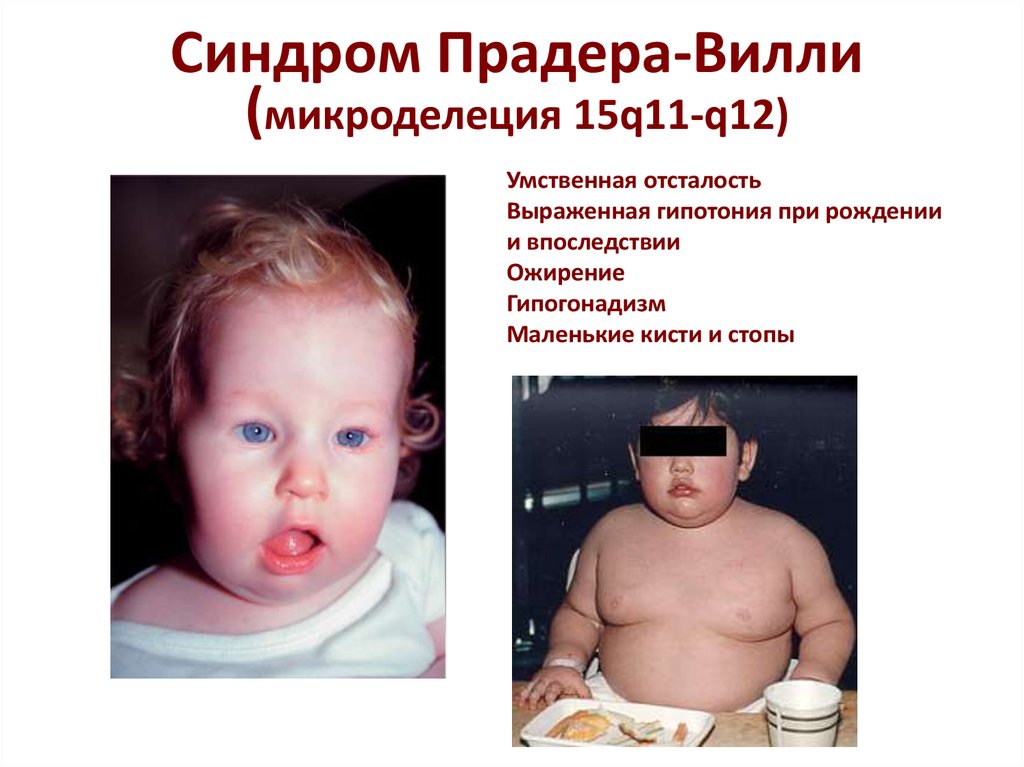
Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г. и встречается у 1 человека на 10000-25000 новорожденных. Заболевание одинаково часто наблюдается у мальчиков и девочек и является основной генетически обусловленной причиной ожирения у лиц старше 1 года. Национальность и раса на распространенность заболевания не влияют. Причиной данного генетического заболевания является отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15 отцовской хромосоме. Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы.
Развитие синдрома Прадера-Вилли всегда связано с хромосомой отцовского происхождения.
Заболевание развивается при:
Делеции (утрате) региона q11-13 отцовской гаметы (выявляется в 70 % случаев).
Полном отсутствии отцовской копии 15-й хромосомы и ее замене дуплицированной материнской копией (материнская дисомия). Наблюдается у 20 % пациентов.
Функциональной дезактивации у плода в результате метилирования (модификации молекулы без изменения нуклеотидной последовательности ДНК) структурно нормального участка q11-13 отцовской хромосомы. Встречается у 5 % больных.
В 2000 г. на основе патогенеза исследователи (Олаецер и др.) выделили три фенотипа синдрома Прадера-Вилли:
- типичный фенотип синдрома Прадера-Вилли, развивающийся при утрате отцовской копии хромосомы;
- более легкий фенотип, отличающийся лучше развитой познавательной функцией;
- выраженный фенотип с большим количеством сердечных патологий.
КОД по МКБ-10
Q87.0
Синдромы врожденных аномалий, проявляющихся преимущественно карликовостью
В подготовке материала использованы сведения из открытых источников.