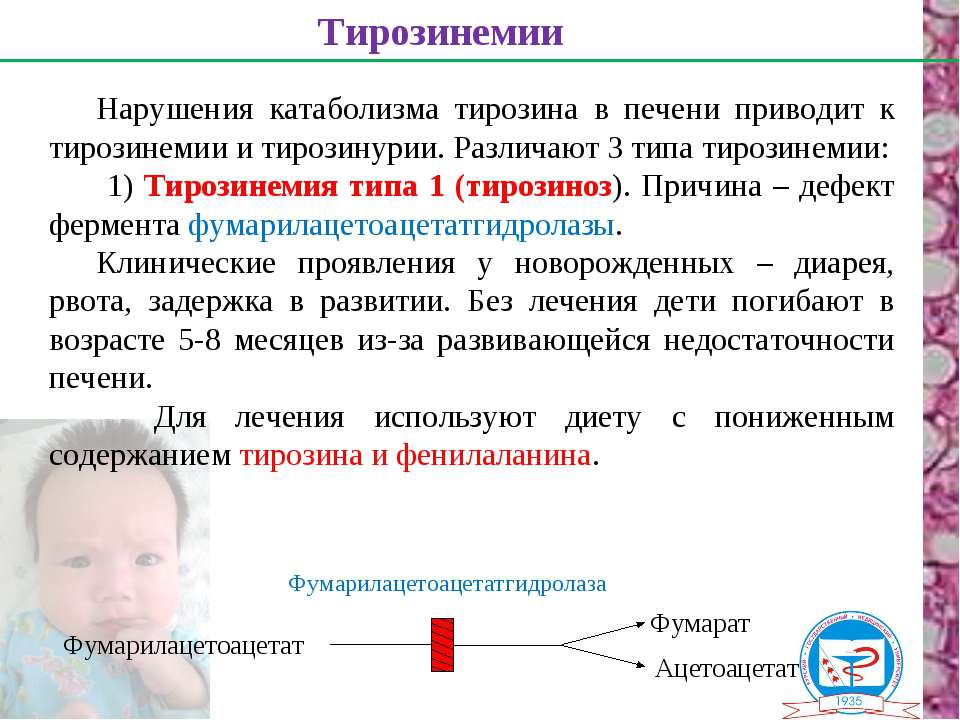
Наследственная тирозинемия 1 типа (НТ-1) – редкое врожденное метаболическое заболевание с аутосомно-рецессивным наследованием, вызванное мутациями гена, отвечающего за фермент фумарилацетоацетатгидролазу (ФАГ).
Этот ген локализован на 15-й хромосоме 15q23-q25. К настоящему времени было выявлено более 40 мутаций гена для данного заболевания, но до сих пор не установлено четкой корреляции между генотипом и фенотипом.
Мутации приводят к нарушению метаболизма тирозина с повреждением печени, почек, периферических нервов. Первым пораженным органом является печень, в течение первых месяцев жизни отмечаются начальные проявления печеночной дисфункции с отдаленным исходом в цирроз и гепатоцеллюлярную карциному (рак печени). Как правило, развивается тяжелый рахит ввиду потери фосфатов. У некоторых пациентов развивается нефрокальциноз и почечная недостаточность.
Возрастание уровня тирозина и метионина в сыворотке вызывают появление «капустного» запаха от больных.
Возраст начала заболевания колеблется от раннего неонатального периода (в этом случае – наиболее быстрое прогрессирование заболевания) до школьного (подросткового) возраста (обычно такой вариант соответствует медленно прогрессирующему поражению печени и почек). До первых проявлений заболевания развитие ребенка не выходит за рамки возрастных показателей.
Наибольшая частота встречаемости НТ-1 зарегистрирована в Квебеке (Канада), где чаще всего заболевание манифестирует прогрессирующей печеночной недостаточностью с первых месяцев, а иногда и дней жизни. В странах Европы течение заболевания чаще приобретает хронический характер с прогрессирующим поражением печени, приводящим к циррозу, и внепеченочными проявлениями, такими как нарушение функции почек, поражение нервной системы, рахит и кардиомиопатия.
Клиническая картина НТ-1 является вариабельной и неспецифичной, что заставляет при постановке диагноза опираться на комплекс симптомов, клинических признаков и лабораторных анализов. Практически в каждом случае развития НТ-1 имеются: увеличение печени (иногда в сочетании с увеличением селезенки), коагулопатия (патологическое состояние организма, которое обусловлено нарушениями свертываемости крови), дисфункция почечных канальцев и рахит.
Частота НТ1 в различных популяциях колеблется от 1:100-120 тыс. живых новорожденных. Частота носительства мутаций НТ1 в популяциях 1:150-100 человек.
Половых различий ни в частоте встречаемости, ни в тяжести течения НТ1 не выявлено. Четких взаимосвязей между генотипом и фенотипом не установлено, различные клинические варианты могут присутствовать у членов одной семьи с одинаковыми мутациями.
КОД МКБ -10
E 70.2
В подготовке материала использованы сведения из открытых источников.