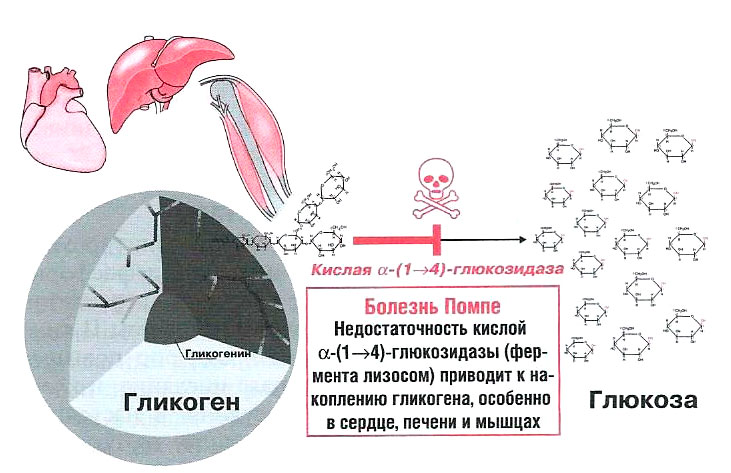
Болезнь Помпе (недостаточность кислой α-1.4-глюкозидазы, или гликогеноз типа II) – тяжелое, прогрессирующее, смертельно опасное нервно-мышечное заболевание с системными проявлениями и разной скоростью прогрессирования.
Болезнь Помпе (БП) относится к редким мультисистемным наследственным болезням накопления, связанным с дефицитом фермента кислой мальтазы (кислой альфа-глюкозидазы) в лизосомах. Преимущественное накопление гликогена отмечено в скелетных мышцах, но в разной степени может обнаруживаться и в других органах и тканях, включая сердечную мышцу, печень, нервную систему, гладкую мускулатуру и т.п.
БП относится к лизосомным болезням накопления (ЛБН). Кроме того, БП можно классифицировать и как нейро-мышечное заболевание, метаболическую миопатию и гликогеноз (БП является единственной патологией, при которой нарушение распада гликогена связано с дефектом функции лизосом), а младенческая форма болезни также относится к кардиологической патологии в связи с выраженными изменениями сердца.
Неонатальная форма заболевания, проявляющаяся сердечной, дыхательной и печеночной недостаточностью, имеет неблагоприятный прогноз. Поздние, медленно прогрессирующие хронические формы, проявляющиеся в возрасте после 1 года жизни с преимущественным поражением скелетной и дыхательной мускулатуры, имеют более благоприятное течение.
Заболевание впервые описано в 1932 г. голландским патологоанатомом Помпе, по имени которого заболевание получило свое название. В 1963 г. биохимик Эром показал, что в основе нарушения метаболизма при болезни Помпе лежит лизосомальная ферментативная недостаточность кислой α‑глюкозидазы (GAA, от англ. α‑glucosidase acid). В1969 г. было доказано существование и поздней формы заболевания, характеризующейся медленно прогрессирующим течением с преимущественным вовлечением скелетных и дыхательных мышц. В последние годы достигнуты значительные успехи в понимании патофизиологических механизмов болезни Помпе, а также в применении заместительной энзимотерапии (ЗЭТ).
Болезнь наследуется по аутосомно-рецессивному типу и встречается у обоих полов с одинаковой частотой. Мутация, ответственная за развитие болезни Помпе, располагается в гене GAA, локализованном на 17q25.2-25 хромосоме. Она была расшифрована в 1989 г. и состоит из 20 экзонов, кодирующих последовательность из 952 аминокислот.
Сегодня идентифицировано более 350 мутаций гена и их число постоянно растет. Хотя некоторые мутации гена GAA встречаются преимущественно в определенной этнической группе, для большинства популяций отсутствуют так называемые founder-мутации, составляющие сколько-нибудь значимую долю всех мутаций популяции и потому целесообразные для прицельного выявления.
Заболевание моногенное, наследуется по аутосомно-рецессивному типу.
При болезни Помпе, независимо от формы, гликоген может накапливаться практически в любых тканях, но при этом имеется преимущественное скопление гликогена в разных органах и тканях, которое уже зависит от формы болезни. Так, при форме МБП (младенческая Болезнь Помпе) гликоген накапливается в скелетной мускулатуре, сердечной мышце, печени, мышцах языка. Реже аномальные отложения гликогена могут встречаться в мышечном слое сосудистой стенки, определяя развитие аневризм и мальформаций, а также – в клетках центральной и периферической нервной системы.
При БППН (Болезнь Помпе с поздним началом), в отличие от МБП, больше всего страдает скелетная мускулатура, в то время как поражение остальных органов и тканей встречается значительно реже и по тяжести поражения не сопоставимо с МБП. На далеко зашедших стадиях БППН снижение слуха, нарушение мочеиспускания и дефекации, а также случайно выявляемые внутримозговые сосудистые мальформации чаще всего являются проявлениями основного генетического дефекта.
Вторичные факторы могут влиять на клиническое течение БП у пациентов с одинаковыми генетическими мутациями, что соответствует слабой корреляции между генотипом и фенотипом. Несмотря на разнообразие клинической картины, БП характеризуется неуклонно прогрессирующим течением с разными вариантами прогрессирования.
Болезнь Помпе – очень редкое заболевание, частота составляет примерно 1 случай на 140 000 (классическая инфантильная форма) и 1 на 60 000 живых новорожденных для взрослой формы. Заболевание встречается практически во всех этнических группах.
КОД по МКБ-10
Е74.0 – Болезни накопления гликогена
Сердечный гликогеноз
Болезнь: Помпе
Недостаточность фосфорилазы печени
В подготовке материала использованы сведения из открытых источников.