Причиной возникновения болезни Фабри являются мутации гена GLA, контролирующего структуру α-галактозидазы А (GLA; EC 3.2.1.22). Ген α-галактозидазы А картирован на длинном плече хромосомы Хq 22.1.
К настоящему времени идентифицировано 599 мутаций и полиморфизмов в гене GLA, в том числе 435 патогенетических «точковых» мутаций, изменяющих кинетические свойства и стабильность галактозидазы А.
Большинство мутаций являются уникальными для каждой семьи. Наиболее часто встречаемые: R227Q и R227X. Обычно больные наследуют дефектный ген от одного из родителей, но около 5% случаев связаны с так называемыми мутациями de novo.
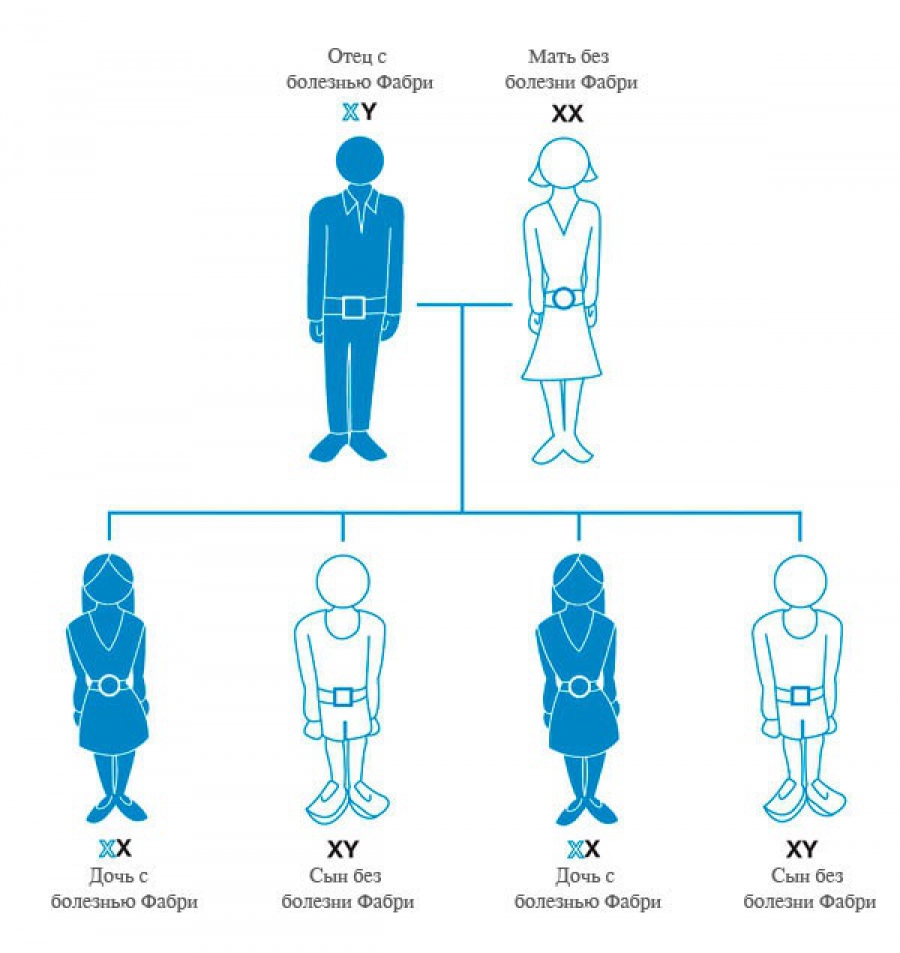
Болезнь Фабри наследуется по рецессивному X-сцепленному типу. Гемизиготные мужчины имеют единственную мутантную Х-хромосому, что определяет классический фенотип болезни. Они передают мутантную хромосому только своим дочерям, но не сыновьям. Таким образом дочери больных болезнью Фабри отцов имеют одну нормальную и одну мутантную хромосому, т.е. являются гетерозиготами. Оставаясь, как правило, клинически здоровыми, они могут передать мутантную хромосому и, следовательно, патологический аллель половине своих потомков. Течение болезни у них, как правило, умеренно-выраженное с более поздним началом, медленным прогрессированием и легкими клинико-патологическими изменениями.
Вместе с тем было показано, что у части гетерозиготных женщины с мутацией гена α-галактозидазы А развиваются тяжелые проявления болезни Фабри, требующие медицинской помощи и вмешательства. Механизм, посредством которого у гетерозиготных женщин развиваются жизнеугрожающие симптомы, неизвестен. У большинства из них имеется почти нормальный уровень циркулирующего фермента за счет того, что случайный процесс инактивации Х-хромосомы (лайонизация) приводит к образованию как дефицитных, так и нормальных клеток. Таким образом, гетерозиготных женщин не следует называть носителями, поскольку носительство подразумевает отсутствие клинических проявлений болезни Фабри.
Гликосфинголипиды, такие, как глоботриаозилцерамид и галабиозилцерамид, имеют терминальный остаток α-галактозила, который отщепляется ферментом α-галактозидазой А. Таким образом, недостаточность или отсутствие фермента приводит к накоплению различных гликосфинголипидов с терминальным α-галактозил остатком.