Болезнь Фабри или болезнь Андерсона-Фабри – наследственное заболевание, относящееся к группе лизосомных болезней накопления, обусловленное значительным снижением активности или отсутствием фермента α-галактозидазы А. Дефицит фермента приводит к накоплению глоботриаозилцерамида и родственных гликофосфолипидов в лизосомах клеток различных органов, включая сердце, почки, нервную систему и эндотелий сосудов.
Болезнь впервые описана в 1898 г. английским дерматологом Андерсоном и немецким дерматологом Фабри. В 1898 году Фабри описал 13-летнего мальчика с нодулярной пурпурой, у которого впоследствии развилась альбуминурия. В том же году Андерсон описал 39-летнего мужчину с ангиокератомой, протеинурией, деформациями пальцев рук, варикозным расширением вен и лимфатическим отеком.
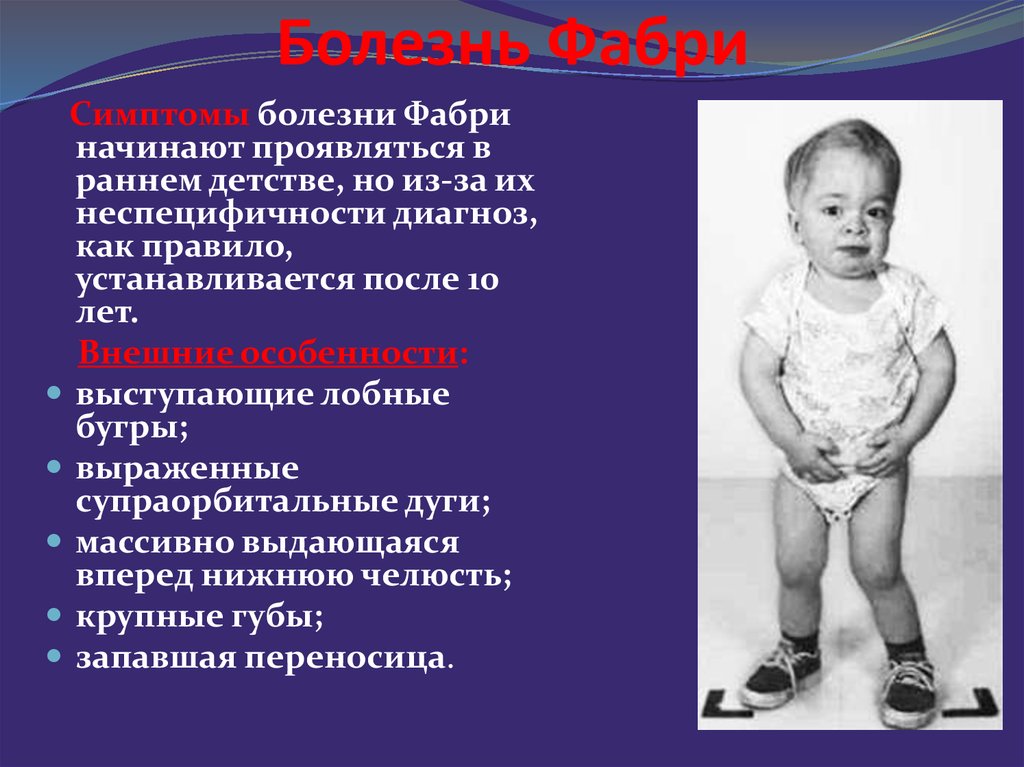
В детском возрасте заболевание проявляется болями в кистях и стопах, ангиокератомами, гипогидрозом, астенией; в более старшем возрасте присоединяются боль в животе, поражение почек, сердца, возможны транзиторные ишемические атаки, инсульт.
Заболевание носит прогрессирующий характер, сопровождается снижением качества и продолжительности жизни. Смерть пациентов, как правило, наступает на 4-м десятилетии жизни от сердечно-сосудистых, цереброваскулярных осложнений или почечной недостаточности.
Болезнь Фабри относится к редким заболеваниям. Распространенность болезни в различных странах мира варьирует в широких пределах (от 1 на 117000 до 1 на 476000 населения).
КОД по МКБ-10
Е75.2 – Другие сфинголипидозы:
-Фабри
-Гоше
-Краббе
-Нимана-Пика
-Синдром Фарбера
-Метахроматическая лейкодистрофия
-Недостаточность сульфатазы
В подготовке материала использованы сведения из открытых источников.